DCS Holdningspapir
Familiær hyperkolesterolæmi (FH)
Udarbejdet af en arbejdsgruppe nedsat af
Dansk Cardiologisk Selskab (DCS).
Arbejdsgruppens medlemmer (alfabe-
tisk):
Lia E. Bang
Rigshospitalet
Henning Bundgaard
Rigshospitalet
Finn Lund Henriksen
Odense Universitetshospital
Henrik Kjærulf Jensen
Aarhus Universitetshospital Skejby
Ib Christian Klausen
Regionshospitalet Viborg
Mogens Lytken Larsen
Odense Universitetshospital
Bent Raungaard
(Formand)
Aalborg Sygehus, Aarhus Universitetsho-
spital
Erik Berg Schmidt
Aalborg Sygehus, Aarhus Universitetsho-
spital
Kristian Thomsen
Sydvestjysk Sygehus Esbjerg
Anbefalingerne er i overensstemmelse med
ESC’s nyligt publicerede rekommandationer
vedrørende håndtering af dyslipidæmi.
let 5-10 mmol/l – hos homozygote endnu
højere.
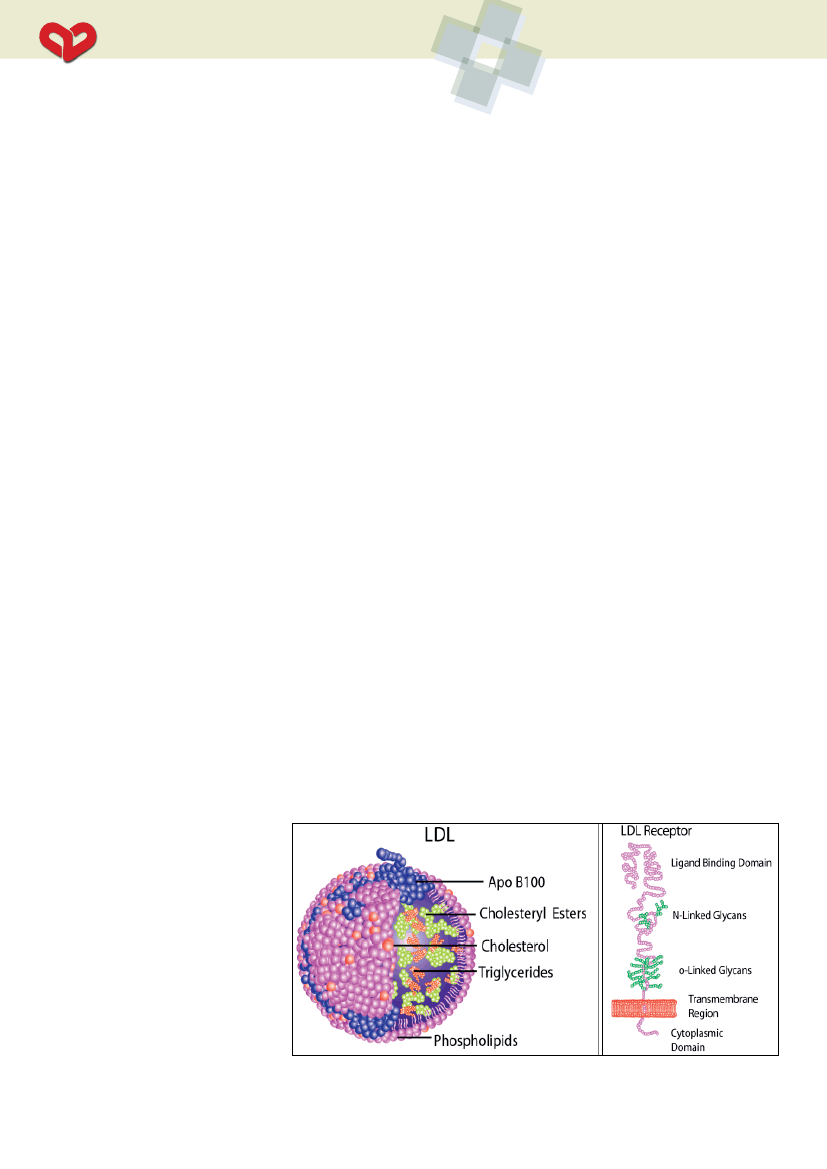
Årsagen til LDL-kolesterolforhøjelsen
er, at den kataboliske kapacitet for LDL-ko-
lesterol er reduceret. Dette medfører øget
koncentration af LDL-kolesterol i blodet. Det
forhøjede LDL-kolesterol skyldes oftest (ca.
90% af tilfældene) defekte LDL-receptorer
betinget af en LDL-receptor gen-mutati-
on, men årsagen kan også være mutati-
onsbetinget defekt i LDL-receptorliganden
apolipoprotein B (apoB) (ca. 5-10% af til-
fældene) (se figur). FH kan også skyldes mu-
tationer i proprotein convertase subtilisin/
Kexin 9 (PCSK 9) genet, hvorved nedbryd-
ningen af LDL-receptorer øges. Der formo-
des at være andre, endnu ikke erkendte
gen-ændringer. Det genetiske baggrundsbil-
lede er således komplekst, og alene i LDL-
receptor genet kendes over 1600 forskellige
mutationer.
Kommissorium
Familiær hyperkolesterolæmi (FH) er en
hyppig, autosomal, monogen arvelig form
for hyperkolesterolæmi, som rammer ca.
1 ud af 500 danskere, og som ubehandlet
medfører betydelig risiko for tidlig iskæmisk
hjertekarsygdom og død. Der ønskes en kort
beskrivelse af tilstanden, herunder dennes
epidemiologi og patofysiologi. Der ønskes
redegjort for 1) retningslinier for diagnostik,
herunder diagnostiske kriterier og undersø-
gelsesmetoder – klinisk og genetisk, 2) fami-
lieudredning, 3) behandling, herunder anbe-
falinger til behandlingsmål, 4) håndtering af
specielle grupper, herunder børn og gravide,
og 5) hvorledes den nationale organisering
af familieopsporing og opfølgning vil kunne
optimeres. Holdningspapiret ønskes udar-
bejdet efter DCS’ retningslinier.
Hyppighed og arvegang
FH er en autosomal dominant arvelig syg-
dom. Den estimerede prævalens af hete-
rozygot FH er ca. 0,2% (1:500) i de fleste
befolkninger herunder også den danske. I
Danmark er der således ca. 10.000 perso-
ner med heterozygot FH. Homozygot FH
forventes hos én pr. million individer. Kole-
sterolomsætningsdefekten er tilstede ved
fødslen.
DCS’ anbefalinger
Præsymptomatisk behandling af FH re-
ducerer morbiditeten og mortaliteten af
aterosklerotisk hjertekarsygdom betydeligt.
For at udnytte den tidlige behandling bedst
muligt, har DCS’ arbejdsgruppe i dette hold-
ningspapir understreget følgende:
At diagnosen FH – eller sandsynlig FH –
er klart defineret og i klinikken relativt
enkel at adskille fra øvrige dyslipidæmier.
At behandlingsmålene ved FH skærpes
i overensstemmelse med ESC’s seneste
anbefalinger, dvs til plasma LDL koleste-
rol < 2,5 mmol/l (<1,8 mmol/l hos højri-
siko-patienter).
At behandling af børn og gravide/am-
mende kvinder præciseres.
At patienten med diagnosen FH – eller
sandsynlig FH – henvises til lipidklinik/
center for arvelig hjertesygdom eller an-
den part med ekspertise i familieopspo-
ring.
At familieopsporingen systematiseres og
udbygges mest muligt for der igennem
at identificere flest mulige patienter med
FH før udvikling af manifest aterosklero-
tisk hjertekarsygdom.
Introduktion
Familiær hyperkolesterolæmi (FH) omfatter
en gruppe genetisk betingede sygdomme,
som er forbundet med svært forhøjet plas-
ma kolesterolniveau og med høj risiko for
udvikling af tidlig aterosklerotisk sygdom.
Det er specifikt niveauet af lav-densitets
lipoprotein kolesterol (LDL-kolesterol), der
er højt. Således har afficerede individer ty-
pisk et LDL-kolesterolniveau, der er dobbelt
så højt som deres ikke afficerede søskende.
LDL-kolesterolniveauet ligger hos hetero-
zygote afficerede voksne typisk i interval-
Betydning af forhøjet LDL
kolesterolniveau
Eksponeringen af arterievæggene for de
høje LDL-kolesterolniveauer fra barndom-
Figur. Illustration af LDL-partiklen og LDL-receptoren.
Familiær hyperkolesterolæmi (FH)
•
1
•
DCS Holdningspapir, april 2012