Udvalget vedrørende Grønlandske Forhold 2008-09
UGF Alm.del Bilag 100
Offentligt
Subscriber access provided by LUNDS UNIV
Article
U, Pu, and Am Nuclear Signatures of the Thule Hydrogen Bomb DebrisMats Eriksson, Patric Lindahl, Per Roos, Henning Dahlgaard, and Elis HolmEnviron. Sci. Technol.,2008,42 (13), 4717-4722• DOI: 10.1021/es800203f • Publication Date (Web): 05 June 2008Downloaded from http://pubs.acs.org on May 18, 2009
More About This ArticleAdditional resources and features associated with this article are available within the HTML version:••••Supporting InformationAccess to high resolution figuresLinks to articles and content related to this articleCopyright permission to reproduce figures and/or text from this article
Environmental Science & Technology is published by the American ChemicalSociety. 1155 Sixteenth Street N.W., Washington, DC 20036
Environ. Sci. Technol.2008,42,4717–4722
U, Pu, and Am Nuclear Signaturesof the Thule Hydrogen Bomb DebrisM A T S E R I K S S O N , *,†,‡P A T R I C L I N D A H L ,§,⊥P E R R O O S ,‡H E N N I N G D A H L G A A R D ,‡,�A N DE L I S H O L M§,#IAEA-MEL, 4 Quai Antoine 1er, MC 98000 Monaco, Monaco,Risoe National Laboratory, P.O. Box 49, DK-4000 Roskilde,Denmark, and Department of Radiation Physics, LundUniversity, SE-221 85 Lund, Sweden
IntroductionIn January 1968, a B52 airplane crashed on the sea ice about12 km west of the Thule air base, NW Greenland. The aircraftwas carrying four hydrogen nuclear weapons. The impacttriggered the conventional explosives within the weaponsand the explosion pulverized the fissile material in the bombs.The debris was scattered around the point of impact andsome square kilometers of the ice was contaminated. Thebenthic marine environment received the fraction of theweapon material that was not recovered from the ice duringthe clean-up operation following the accident and probablyalso a fraction injected during the accident, as the impactcaused part of the ice to break up (1–4).Until now the source term and characteristics of the fourdisintegrated weapons have been poorly described in theopen literature. The source term is an important aspect toknow when dose assessments and environmental impactstudies are to be conducted in a contaminated area. For thefirst 30 years, all studies were focusing on the increased levelsof Pu and Am in the area as well as the transfer of theseelements into the Arctic food web. The fissile material in theweapons were assumed to be a plutonium device and noinvestigations concerning uranium can be found. Later, someinvestigations using ICP-MS and high-resolutionRspec-trometry for the determination of the240Pu/239Pu isotopicratio suggest that the debris consists of different Pu sources(5–8). However, since the ratios were determined on bulksediment samples, the ratios could represent a mixed ratioof different sources and the uniqueness from each sourcecould not be determined. To identify the characteristicnuclear fingerprints in such areas, one should study singleisolated hot particles, which most probably are fragments ofthe weapon material and consist of nuclides from one singlesource.It took 31 years before the first report of uranium in theThule accident was published (9). This hot particle studyshowed that Pu and U coexisted in the weapon material.One explanation that U was not reported earlier may be thatall analyses were conducted on bulk sediment from the area.It is very easy to study Pu originating from the accident inthese sediments as the levels are much higher than the Pufallout levels. For U it is the opposite; the additionalcontribution from the accident might have been somekilogram of high enriched uranium spread out on somesquare kilometers of the sea floor and one can estimate thatin the top 10 cm of the sediment in such an area there existabout 100 kg of natural uranium, so the accident U isotopicsignature would to a great extent be blurred out with theisotopic signature of natural U.Studies on isolated particles from the Thule sediment haveshown higher235U/238U ratios than the natural U isotopiccomposition (10,11).In a recent study using secondary ionmass spectrometry (SIMS) to study single hot particles (10),it was shown that the Thule particles consist of differentweapon-grade Pu and highly enriched U isotopic composi-tions; however, no U/Pu ratios were reported. One of thechallenges of working with hot particles are the determinationof the U isotopic composition, as these particles typicallycontain a tenth of a nanogram of uranium, and these amountsare typically procedure blank values of U conducted in normallaboratories, so the radiochemistry must be conducted underultrapure conditions, preferably in clean laboratories.In the present study the complete isotopic signature andthe age of the nuclear material involved in the Thule accidentare determined. It focuses on the actinides, U, Pu, and AmVOL. 42, NO. 13, 2008 / ENVIRONMENTAL SCIENCE & TECHNOLOGY9
Received January 25, 2008. Revised manuscript receivedApril 8, 2008. Accepted April 15, 2008.
This study concerns an arctic marine environment that wascontaminated by actinide elements after a nuclear accident in1968, the so-called Thule accident. In this study we haveanalyzed five isolated hot particles as well as sediment samplescontaining particles from the weapon material for thedetermination of the nuclear fingerprint of the accident. Wereport that the fissile material in the hydrogen weapons involvedin the Thule accident was a mixture of highly enricheduranium and weapon-grade plutonium and that the mainfissile material was235U (about 4 times more than the mass of239Pu). In the five hot particles examined, the measureduranium atomic ratio was235U/238U)1.02(0.16 and the Pu-isotopic ratios were as follows:240Pu/239Pu)0.0551(0.0008 (atom ratio),238Pu/239+240Pu)0.0161(0.0005 (activityratio),241Pu/239+240Pu)0.87(0.12 (activity ratio), and241Am/239+240Pu)0.169(0.005 (activity ratio) (reference date2001-10-01). From the activity ratios of241Pu/241Am, weestimated the time of production of this weapon material tobe from the late 1950s to the early 1960s. The results fromreanalyzed bulk sediment samples showed the presence of morethan one Pu source involved in the accident, confirmingearlier studies. The238Pu/239+240Pu activity ratio and the240Pu/239Pu atomic ratio were divided into at least two Pu-isotopic ratiogroups. For both Pu-isotopic ratios, one ratio group hadidentical ratios as the five hot particles described above andfor the other groups the Pu isotopic ratios were lower (238Pu/239+240Pu activity ratio∼0.01 and the240Pu/239Pu atomic ratio∼0.03). On the studied particles we observed that the U/Puratio decreased as a function of the time these particles werepresent in the sediment. We hypothesis that the decrease inthe ratio is due to a preferential leaching of U relative to Pu fromthe particle matrix.
* Corresponding author phone:+37797977222; fax:+37797977273;e-mail: [email protected].†IAEA-MEL.‡Risoe National Laboratory.§Lund University.⊥Current address: EC-JRC-IRMM, Retieseweg 111, B-2440 Geel,Belgium.#Current address: Centro Nacional de Aceleradores, Avda/Thomas Alba Edison, no. 7, 41092 Sevilla, Spain.�Dr. Henning Dahlgaard: 1950-2005.10.1021/es800203f CCC: $40.752008 American Chemical Society
4717
Published on Web 06/05/2008
in five isolated particles that have been totally dissolved underultraclean conditions to ensure no bias of the235U/238U ratio.In addition, retrospective analyses of bulk samples, wherethe activity most probably originated from one single particle,completed the determination of the Pu isotopic ratios (240Pu/239Pu and238Pu/239+240Pu).
Experimental SectionThe particles were separated from the top sediment (0-9cm). The sediments were sampled by sediment corers closeto the point of impact of the Thule accident, during thesampling expeditions to Thule in 1979, 1984, and 1997 (4,6).The particle separation technique has been described in detailelsewhere (12). Briefly, the technique is based on samplesplitting, using the gamma emitting (59.54 keV,nγ)35.9%)241Pu daughter241Am, as an indicator of hot particles. Thesplitting of the samples continued until only a few grainsremained, including the hot particle. The hot particles werefurther identified and localized using real-time digital imagingsystems.Dissolution of (U,Pu)O2hot particles is notoriously difficultusing mineral acids (13). We checked that complete dis-solution of the particles had occurred by comparing thegamma activity, measured by a high-purity germanium(HPGe) detector on the whole particle, with theRactivity of241Am determined after dissolution and chemical separation.Different analytical methods were used to determine theelemental composition, activity, and mass of U, Pu, and Amin each particle. High-resolution inductively coupled plasmamass spectrometry (HR-ICP-MS) and alpha spectroscopywere used to determine the isotopic and element ratios andthe masses and activities in the particles. The instrumentsused were a Micromass PlasmaTrace2 high-resolution ICP-MS with an x-flow nebulizer and passivated implanted planarsilicon (PIPS)Rdetectors.The particles were dissolved in a mixture of hydrofluoricacid and nitric acid (ratio 1:3) in high-pressure Teflon cells.The dissolution procedure began with 0.1 mL of HF and 0.3mL of HNO3, which was evaporated to dryness, and this wasrepeated 30 times. Finally, 1 mL of HF and 3 mL of HNO3were added to the closed Teflon cells, which were then placedon a hot plate so that the mixture was under pressure for24 h. The solution was then evaporated to dryness. Inaddition, one particle (later referred to as TPE) was dissolvedin a microwave oven, CEM MARS 5. A mixture of 7 mL of HF,7 mL of HNO3, and 1.5 mL of HCl was added to the Tefloncell and heated for 20 min in the microwave oven at amaximum pressure of 250 psi (17.2 bar) and a maximumtemperature of 200�C.Twenty-five milliliters of saturatedboric acid was then added and the mixture was heated foranother 20 min in the microwave oven, to break thePu-fluoride complexes formed in the dissolution process.The solutions from both dissolution methods were evapo-rated to dryness and 3 times 0.3 mL of HNO3was added andevaporated to dryness, to ensure that all the fluoridecomplexes were destroyed. The dissolved particles werethereafter diluted in 50 mL of 5% HNO3(Ultra Pure Sea Starquality and>18MΩ-cm H2O). The analysis of Pu and U wasdone on radiochemically separated aliquots of the samplesolution as well as on radiochemically unseparated aliquots.The sizes of the aliquots were 0.2-1.5 mL, depending on theactivity of the particle. Radiochemical separation was per-formed using ion exchange (AG 1×4, 100-200 mesh, BIORAD). To minimize the use of acid, 1.5 mL columns with 0.7mL resin were used. All acid used was of Ultra Pure Sea Starquality, except HF which was of pro analysis Merck quality.Unused Savillex Teflon capsules were acid-washed in boiling12 M HCl before use. The samples were prepared in a cleanlaboratory. These precautions were taken to reduce cross-contamination by naturally occurring uranium isotopes.47189
Subsamples of each dissolved particle solution wereanalyzed with chemical yield determinants as well as without.The chemical yield determinants are added to monitor theloss of a certain element in the chemical separation proce-dures and to determine the mass and activity of eachradionuclide in the sample. As chemical yield determinantswe used233U,242Pu, and243Am. The subsamples analyzedwithout chemical yield determinants were used to determinethe isotopic ratios; this as well as all isotopic chemical yielddeterminants has impurities of the isotopes of interest in thesample and thereby could bias the isotopic ratio determi-nation. In addition, procedure and machine blank sampleswere prepared to assess the background levels.U, Pu, and Am Separation.The subsamples wereevaporated to dryness and thereafter dissolved in 8 M HNO3.Pu(IV) was absorbed onto the columns. The eluate wascollected for Am and U, which was then separated using themethod described by Holm (14). Thorium was hereaftereluted with 9 M HCl, followed by the elution of Pu with NH4I+9 M HCl. The samples were then evaporated to drynessand the samples intended for ICP-MS analysis were dilutedto 3% HNO3solutions (10 mL). The samples measured byRparticle spectrometry were electrodeposited onto stainlesssteel discs in accordance with the method described byHallstadius (15).241Pu Determination.The241Pu was determined by the241Am ingrowths on PuRdiscs. The Pu discs were remeasured6 years after the first measurement (i.e., at the same time aswhen Pu was radiochemically separated from Am). As the241AmRspectra overlaps with the238PuRenergies, thecalculated decay corrected counts from238Pu were subtractedfrom the totalRcounts in these channels.
ResultsParticle Destruction.It is known that a (U,Pu)O2matrix isdifficult to dissolve using mineral acids. Experiments per-formed at Risoe National Laboratory (1) have shown that8-40% of the Pu remains undissolved in bulk Thule sedimentsamples when using the Aqua Regia leaching techniquedescribed by Talvitie (16). Incomplete dissolution of Thulehot particles have been reported in other studies, confirmingthis (11,17).An alternative method for particle dissolutioncould be the use of fusion agents as KF and borate fusionapproach (18,19).However, the reported238U proceduralblank values from this dissolution technique are similar tothe amount expected in the particles (range from 50 to 100ng (19)), and for that reason these procedures are notoptimized for single hot particles studied for the U isotopicsignature. To check if the particles in the present study weredissolved, theγ-raymeasurements of241Am on the undis-solved particle and the determinedRactivity (determinedfrom an aliquot of the dissolved particle solution) werecompared. A good agreement between the241Am activitiesin the particles determined byγandRspectrometry wereachieved (coefficient of determination,R2)0.9991), indicat-ing that the particles were totally dissolved using the methodsdescribed in this article.Plutonium Content.The results from HR-ICP-MS can beseen in Table 1 and fromRspectrometry in Table 2. Theresults from a UKAEA Pu reference material solution (sam-ple number: UK Pu 5/92138) were in good agreement withthe reported values in the certificate (the atomic ratios agreedwithin 0.01%). The mean atom ratio,240Pu/239Pu, for the fiveparticles is 0.0551(0.0008 (1 SD,n)5) and the corre-sponding activity ratio is 0.2057(0.0011 (1 SD,n)5).There is also good agreement between the two differentanalytical techniques, i.e., the HR-ICP-MS andRspectrometryfor the239+240Pu measurements. The mean activity ratio of238Pu/239+240Pu is 0.0161(0.0005 (1 SD,n)5) and241Pu/239+240Pu)0.87(0.12 (1 SD,n)5), and the mean activity
ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 42, NO. 13, 2008
TABLE 1.Pu Isotope Content and Ratios in the Particles on Mass and Activity Basis As Measured by HR-ICP-MSparticle idTPATPBTPCTPDTPEparticle idTPATPBTPCTPDTPE239+240Pu
(ng)
239Pu
(ng)
240Pu
(ng)
240Pu/239Pu
atom ratio
2.51(0.094.37(0.1610.66(0.3928.9(1.132.2(1.2239+240Pu
2.380(0.0884.14(0.1510.10(0.3727.4(1.030.5(1.1239Pu
0.133(0.0060.232(0.0090.559(0.0221.521(0.0601.709(0.067240Pu
0.0544(0.00170.0558(0.00130.0559(0.00110.0554(0.00120.0541(0.0011240Pu/239Pu
(Bq)
(Bq)
(Bq)
activity ratio
6.52(0.2511.35(0.4327.6(1.074.9(2.883.5(3.1
5.41(0.209.41(0.3522.95(0.8462.2(2.369.2(2.5
1.111(0.0461.943(0.0774.69(0.1812.75(0.5014.32(0.56
0.2056(0.00650.2065(0.00490.2042(0.00410.2050(0.00440.2071(0.0041
TABLE 2.Pu Isotope Content and Ratios in the Particles on Activity Basis As Measured byr-Spectrometry(Reference Date:2001-10-01)238Pu/239+240Pu241Pu/239+240Pu241Am/239+240Pu
particle idTPATPBTPCTPDTPE
239+240Pu
(Bq)
activity ratio0.0167(0.00100.0165(0.00090.0158(0.00080.0156(0.00080.0158(0.0007
activity ratio0.75(0.150.84(0.131.06(0.120.83(0.110.86(0.13
activity ratio0.173(0.0100.172(0.0090.162(0.0080.172(0.0090.166(0.009
age (years);determined byingrowths43.8(541.6(336.3(441.8(2.440.4(3
6.22(0.239.78(0.3526.38(0.9474.6(2.884.1(3.0
TABLE 3.Uranium in the Particles Measured by HR-ICP-MSparticle idTPATPBTPCTPDTPE235U
(ng)
238U
(ng)
235U/238U
atom ratio
235U/239Pu
mass ratio
sampling date1979-8-231984-8-111984-8-111979-8-231997-8-25
9.28(0.6310.00(0.6813.98(0.9574.65(5.0634.75(2.36
8.34(0.5712.02(0.8216.29(1.1165.34(4.4430.27(2.07
1.112(0.0050.832(0.0030.858(0.0041.143(0.0041.148(0.006
3.90(0.302.41(0.191.38(0.112.73(0.211.14(0.09
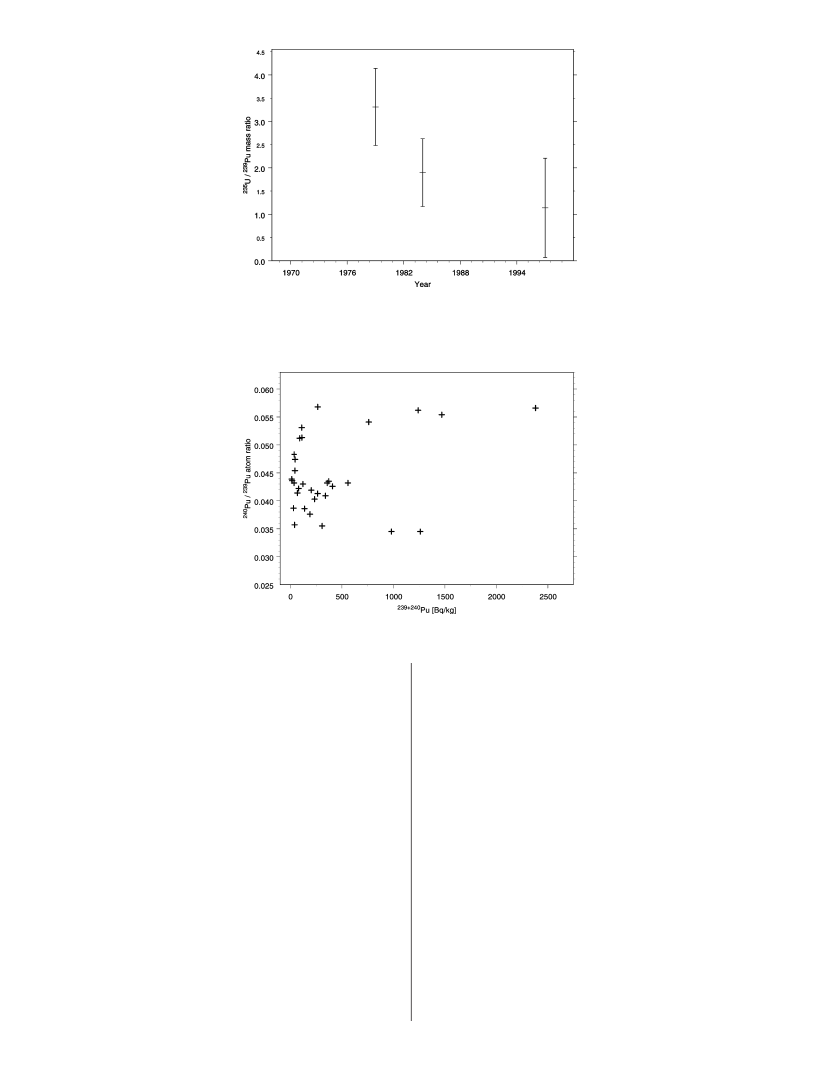
ratio241Am/239+240Pu is 0.169(0.005 (1 SD,n)5) in the 5particles, reference date: 2001-10-01.Uranium Content.The uranium results from the HR-ICP-MS measurements are shown in Table 3. These resultsshow similar isotopic ratios for the five particles,235U/238U)1.02(0.16 (1 SD,n)5) which is equivalent to about 50%mass enrichment of235U. The slightly lower ratio can be aneffect of some cross-contamination of natural uranium, whichhas a much lower ratio. However, these samples wereradiochemically separated and prepared in a clean laboratoryand for the sub samples where the235U/238U ratio wasdetermined no radiochemical yield determinant (233U) wasused, to minimize cross-contamination of uranium.The mass ratio of the fissile material in the particles,235U/239Pu, is less affected by natural uranium cross-contaminationthan the235+238U/239+240Pu ratio (Table 3). It appears that theratio decreases with the time the particle has been exposedto the marine sediment (Figure 1). We believe that the resultreflects preferential leaching of the uranium from theparticles.
DiscussionsThe published investigations concerning the Thule accidenthave historically been focused on plutonium and very littleon uranium in the contaminated area. The Pu inventoriesand Pu and Am transfer in the food web have been the maininterest in these studies (2–4). The source term was deter-mined on bulk samples withRspectrometry and character-
ized with the isotopic signature of238Pu/239+240Pu ratio and241Am/239+240Pu ratio.It took several years before the first240Pu/239Pu ratios werepublished (5–8,20, 21).No unique values have been foundin these studies as the240Pu/239Pu isotopic ratios have beenscattered (ranging from 0.022 to 0.059 atom ratio), whichsuggests that there is more than one unique source term ofthe Thule accident debris. These ratios were determined onbulk sediments and could be mixed ratios from differentsources and the fingerprint from each source is then difficultto determine. Dahlgaard et al. (6) measured the240Pu/239Puatom ratios by HR-ICP-MS on 32 bulk sediment samplescollected on the 1997 sampling expedition to Thule andplotted the ratio versus the activity in the analyzed sample.It is clear from Figure 2 (redrawn from Dahlgaard et al. (6))that the high-activity samples are split into two groups, withmass ratios of 0.057 and 0.035. The mean ratio of thesemeasurements is 0.0442(0.0073. There is an indication ofa group around the mass ratio of 0.043. However, these aresamples from locations close to the point of impact, lessthan 1 km, and they are considered as low active for theselocations, and possibly, they could be a mixture of the othersources and reflect the mean ratio between the sources ratherthan a group itself. However, that it should be a ratio group,rather than a mean ratio, is supported by the same Pu isotopicratio found in one hot particle analyzed by Ranebo et al.(10).To study the nuclear signatures of the sources present,single hot particles should be analyzed, which most probablyVOL. 42, NO. 13, 2008 / ENVIRONMENTAL SCIENCE & TECHNOLOGY9
4719
FIGURE 1. Mean mass ratio,235U/239Pu, in particles collected during a specific year. There is a tendency that the uranium content islower in the particles exposed in the marine sediment environment for a longer time, which would mean that there is preferentialleaching of uranium from the particle in the sediment. The standard deviation for the ratio in 1997 is the square root of the ratio, asonly one particle has been analyzed from this year; the standard deviation from the HR-ICP-MS measurement is much less than thatshown (see Table 3).
FIGURE 2. Graph showing the240Pu/239Pu atom ratio in bulk sediment samples sampled in 1997. The atom ratios are grouped into twogroups around 0.056 and 0.035 and a possible group around 0.043. Data from Dahlgaard et al. (6).are fragments of the weapon components, i.e., originatingfrom one single source. The results from the particles studiedherein indicate only one single source with the isotopic ratios:238Pu/239+240Pu activity ratio of 0.0161(0.0005,n)5, andthe240Pu/239Pu atom ratio of 0.0551(0.0008,n)5. The240Pu/239Pu ratios are equal to the ratios observed byDahlgaard et al. (6), Sturup et al. (7), and Mitchell et al. (8)�in their most active bulk sample. Lind et al. (11) analyzedthree Thule hot particles, all with equal240Pu/239Pu atomratios as the particles investigated in this study. Ranebo etal. (10) studied eight Thule hot particles by SIMS and foundfour different240Pu/239Pu atom ratios. Five of these had a240Pu/239Pu atom ratio similar to those of the particles in thisstudy (around 0.058) and the other three particles had theratios 0.042, 0.036, and 0.028, respectively. Concluding the240Pu/239Pu isotopic atom ratio discussion, four characteristicgroups have been identified from hot particle studies withthe ratios of 0.028, 0.036, 0.042, and 0.056. And it also appearsthat most of the Thule debris originates from the source witha240Pu/239Pu isotopic atom ratio of 0.056 and an238Pu/239+240Puactivity ratio of 0.016. This, as all the five studied hot particlesin this study and most of the hot particles studied by others(10,11),has this ratio and a few other particles with lowerratios have been analyzed. One explanation, excluding thestatistical possibility that the present study only has sampledparticles from the “high Pu isotopic ratio group”, could be47209
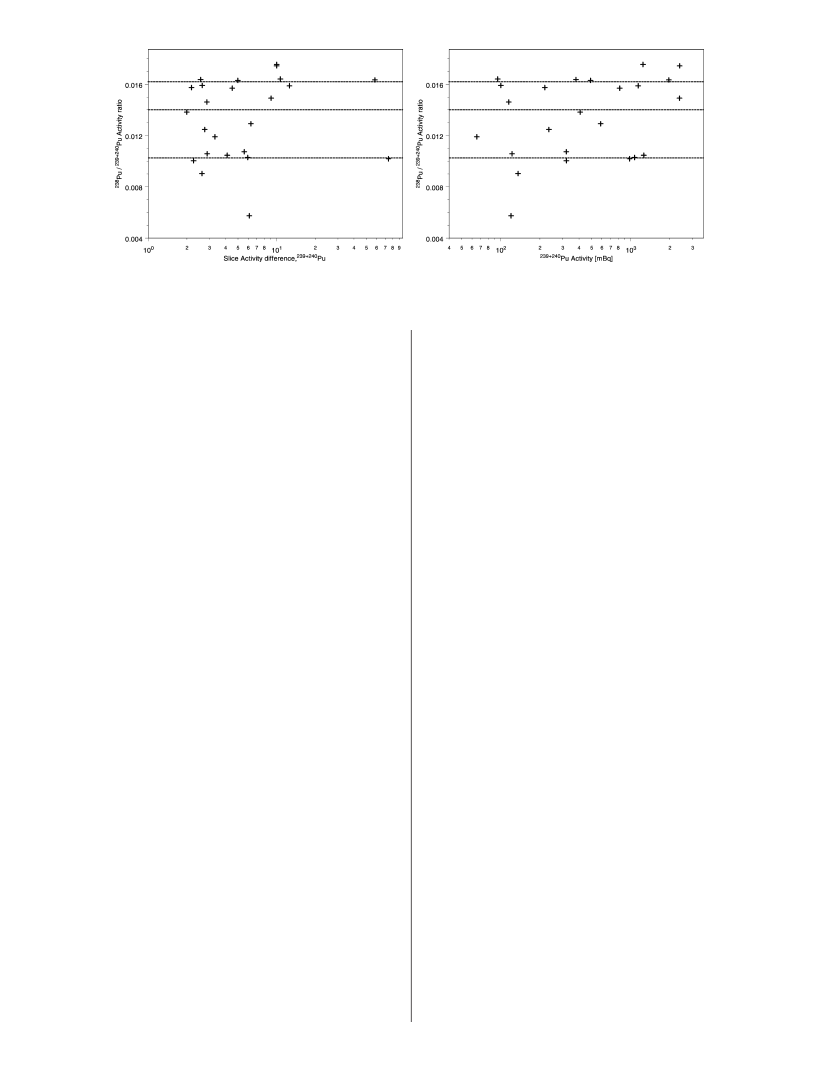
that this source, during disintegration, produced largerparticles. This is also indicated from the bulk sedimentsamples, where the samples with the highest activity originatefrom this source. In addition, larger particles are more easilyidentified and separated than the small sized particles usingthe sampling splitting method. It cannot be determinedwhether the different sources originate from differentweapons or from different parts within the weapons.The activity ratio of238Pu/239+240Pu in Thule sedimentshas been determined in several hundreds of samples overthe past 39 years (2–4,6, 22, 23).These ratios have beenmeasured byRspectrometry performed on bulk sedimentsamples (0.5-5 g). The mean238Pu/239+240Pu ratio from thesediments collected during the Thule 1984, 1991, and 1997expeditions is 0.014(0.004 (n)328), decay corrected to2001. This mean ratio is derived from the sediments close tothe point of impact (within a radius of 8 km). In this area,the contribution of global fallout Pu (with an activity238Pu/239+240Pu ratio of 0.025) is less than 1% of the Pu inventory.This mean ratio is lower than the ratios found in the fiveparticles studied in the present paper, which support thetheory that more than one unique source term shouldbe present. However, since the plutonium activity in thesediment is, to a great extent, associated with hot particles(1), and high-activity samples have been analyzed, it couldbe possible to investigate the unique ratios of the sources
ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 42, NO. 13, 2008
FIGURE 3. Two plots showing the238Pu/239+240Pu activity ratios in bulk sediment samples. To the left are the ratios plotted versus thedeviation from the mean activity of the activity concentration above and under the slice and to the right are the ratios plotted versusthe activity concentration in the subsample. The ratios are grouped into two groups around 0.016 and 0.010, and there are very fewobservations close to the mean ratio of 0.014.present in the sediment by applying some criteria to olddata. One such criterion is to use the238Pu/239+240Pu ratiosin the subsamples with the highest activities, which are likelyto originate from one single hot particle. The countingstatistics in theRspectra of these samples are also better andso the uncertainty in the238Pu measurements from interferingpeaks originating from the short-lived daughters from thethorium series is lessened. However, as the sediments at thepoint of impact have very high activity concentrations andnumerous particles are present, a second criterion must beapplied. Thus, the activity deviation between a particularsediment slice and the mean activity between the slice overand under this slice must be large; i.e., an abnormal activityconcentration should be observed in the Pu sediment depthprofile. We have performed such a study on the samplescollected at the 1997 sampling expedition to Thule, fulfillingthese two criteria. It is clear from Figure 3 that almost noneof these samples have the calculated mean238Pu/239+240Puratio of 0.014. The238Pu/239+240Pu ratios are grouped more orless around two ratios of 0.016 and 0.010. In the plot to theleft in Figure 3, the ratios are plotted versus the activityconcentration deviation, i.e., the second criterion. The twosamples with the largest deviation most probably originatefrom one single source (hot particle) and they have activityratios of 0.0102 and 0.0163, respectively. In the right-handplot of Figure 3 the ratios are plotted versus the activityconcentration and it is clear that the samples with highestactivity concentrations stretch out around the 0.016 ratiogroup. Assuming that only these two unique238Pu/239+240Puratios are present and the mean ratio observed is 0.014, thiswould mean that two-thirds of the debris originates from thesource with the238Pu/239+240Pu activity ratio of 0.016. Thehigh238Pu/239+240Pu ratio is in good agreement with the fivehot particles studied in the present paper. These particles allhave higher activity (6-84 Bq239+240Pu) than the analyzedbulk sediment samples, where the highest activities of thesamples were about 2.5 Bq239+240Pu.The241Pu activity of the particles was calculated by241Amingrowths on PuRdisks. The241Pu/239+240Pu can be seen inTable 2. The mean activity ratio for the five particles was 0.87(0.12 in October 2001. In Table 2 the calculated age (timesince Pu was chemically separated from the Am) of theweapon-grade Pu material can be seen. The ages indicatethat this fission material was produced in the late 1950s andearly 1960s. This is in good agreement with Aarkrog et al. (4)who estimated the time of241Am extraction to be February1960(4 years. Moring et al. (17) estimated a younger age(1967(3 years) of the material calculated in one hot particle.241Am/239+240Pu ratios have also been determined severaltimes in Thule sediments (4,6, 22–24).However, the ratioshave in some cases been determined on different aliquots,which often is the case in the analysis of high-activity samples.Therefore, the ratios determined are of no use in evaluatingthe unique source terms. The ratio is still increasing due tothe decay of241Pu and will reach maximum ratio 73 yearsafter the Am was radiochemically separated from Pu. To beable to compare241Am data sets from different Thule samplingexpeditions, the amount of241Pu and the manufacture dateof the different sources of the weapons must be known. Itis probable that the241Pu content differs between the differentsources in the Thule accident debris, as the241Am/239+240Puactivity ratio reported shows large variations (5,24).If thesources were made at different times, it would be verycomplicated to determine the expected current241Am/239+240Pu activity ratios. The mean ratio of the five particlesis 0.169(0.005 (reference date: December 2001) and thisratio will reach its maximum around the year 2030 and thenstart to decrease to lower ratios.Very few reports on uranium in the Thule debris havebeen published. In the investigated five particles in this studyit is clear that the fissile material in the nuclear weaponsinvolved in the Thule accident is a mixture of highly enrichedU (about 50% mass enrichment of235U) and weapon-gradePu. As only particles originating from one unique sourcehave been analyzed for both U and Pu, this is at least truefor these. It is most probably true also for the other Pu isotopicmixtures in the Thule debris, as no study has yet found aThule particle consisting of only Pu without enricheduranium. Ranebo et al. (10) show that all the particlesanalyzed in the SIMS have high enriched uranium mixedwith the Pu. The235U/238U atom ratio ranged from 0.959 to1.437 in their study. This is in good agreement with the ratiosof the five particles (235U/238U)1.02(0.16) we analyzedwith the ICP-MS technique, also showing that only limitedcross-contamination from natural U was introduced in ourmethod.As shown in Figure 1, the235U/239Pu mass ratio isdecreasing with time, indicating that uranium is leaving theparticle lattice. With the belief that this represents apreferential leaching of uranium compared to plutonium,the estimated mass of235U was around 4 times the mass of239Pu in the fresh accidental debris. Preferential leaching hasbeen demonstrated in several experiments conducted inlaboratories where the leaching/corrosion of UO2fromnuclear fuel has been studied (25,26).In these experiments(conducted in alkaline as well as acid salt solutions of differenttypes) most data indicate a stronger release of uranium thanplutonium. These studies can partly explain the decreasingU/Pu ratio with time, shown in Figure 1. It can be seen inthe figure that the235U/239Pu ratios are associated with largeVOL. 42, NO. 13, 2008 / ENVIRONMENTAL SCIENCE & TECHNOLOGY9
4721
variations for a specific year. The large variance in the ratioscan be explained by the fact that few particles are comparedeach year and also to some extent by the fact that the particlesmove up and down in the sediment as a consequence of thebioturbation combined with the leaching rate probably beinga function of sediment depth, interstitial water chemistry,sediment coating, and particle composition. When consider-ing particle dissolution and potential release of weaponplutonium and uranium, it has also to be considered thatover time most of the particles will reach sediment depthswhere oxic dissolution kinetics may not be applied (e.g., theUO2model by De Pablo et al. (26)), as during oxygen-deficientor anoxic conditions uranium may not become oxidized andthus the particle dissolution rates may be significantlyreduced. Rollin et al. (25) observed UO2dissolution rates�that were a factor of 1000 less under reducing conditionsthan those in oxic conditions with the same bicarbonateconcentrations for both cases. On the other hand, dissolutionrates of plutonium were only reduced by a factor of 10 in thesame experiment.The particles formed during the Thule accident arehowever likely to be rather different compared to the spentUO2fuel. The specific surface area (porosity of particles) isprobably different due to fast cooling after the fire at Thule.The particles formed at Thule may be treated as site-specificparticles and be regarded as a unit of their own. Knowledgeon their dissolution and corrosion rates may be derived onlyfrom experiments conducted with the very same material.
(9)
(10)
(11)
(12)(13)
(14)(15)(16)
AcknowledgmentsThe authors acknowledge NKS framework program BOK-2and the Royal Physiographic Society for financial support.We also acknowledge Dr. D. H. Oughton and Dr. L. Skipperudat the Agricultural University of Norway. The IAEA is gratefulfor the support provided to its Marine Environment Labo-ratories by the Government of the Principality of Monaco.
(17)
(18)
(19)
Literature Cited(1) Eriksson, M. On weapons plutonium in the Arctic environment(Thule, Greenland), Ph.D thesis, Risø-R-1321(EN), Risø NationalLaboratory, Denmark, 2002.(2) Aarkrog, A. Radioecological investigation of plutonium in anarctic marine environment.Health Phys.1971,
20,31–47.(3) Aarkrog, A. Environmental behaviour of plutonium accidentallyreleased at Thule, Greenland.Health Phys.1977,
32,271–284.(4) Aarkrog, A.; Dahlgaard, H.; Nilsson, K.; Holm, E. Further studiesof plutonium and americium at Thule, Greenland.Health Phys.1984,
46,29–44.(5) Dahlgaard, H.; Chen, Q. J.; Sturup, S.; Eriksson, M.; Nielsen,�S. P.; Aarkrog, A. Plutonium isotope ratios in environmentalsamples from Thule (Greenland) and the Techa River (Russia)measured by ICPMS and alpha-spectrometry. InProceedings ofInternational Symposium on Marine Pollution,IAEA-TECDOC-1094, 1998; Monaco, 1999; pp 254-259.(6) Dahlgaard, H.; Eriksson, M.; Ilus, E.; Ryan, T.; McMahon, C. A.;Nielsen, S. P. Plutonium in the marine environment at Thule,NW-Greenland after a nuclear weapons accident. InPlutoniumin the environment;Kudo, A., Ed.; Elsevier Science: Oxford, UK,2001; pp 15-30..(7) Sturup, S.; Dahlgaard, H.; Nielsen, S. C. High resolution�inductively coupled plasma mass spectrometry for the tracedetermination of plutonium isotopes and isotope ratios inenvironmental samples.J. Anal. Atom. Spectrom.1998,
13,1321–1326.(8) Mitchell, P. I.; Leon Vintro, L.; Dahlgaard, H.; Gasc, C.; Sanchez-Cabeza, J. A. Pertubation in the240Pu/239Pu global fallout ratio(20)
(21)
(22)
(23)
(24)
(25)
(26)
in local sediments following the nuclear accidents at Thule(Greenland) and Palomares (Spain).Sci. Total Environ.1997,
202,147–153.Eriksson, M.; Dahlgaard, H.; Ilus, E.; Ryan, T.; Chen, Q. J.; Holm,E.; Nielsen, S. P. Plutonium in the marine environment of ThuleAir Base, N.W. Greenland. Inventories and distribution insediments 29 years after the accident. InExtended abstracts. 4.International conference on environmental radioactivity in theArctic, Edinburgh (GB), 20-23 Sep 1999;Strand, P., Jolle, T.,�Eds.; Norwegian Radiation Protection Authority: Osteras, Swe-den, 1999; pp 60-62.Ranebo, Y.; Eriksson, M.; Tamborini, G.; Niagolova, N.; Bildstein,O.; Betti, M. The Use of SIMS and SEM for the Characterizationof Individual Particles with a Matrix Originating from a NuclearWeapon.Microsc. Microanal.2007,
13,179–190.Lind, O. C.; Salbu, B.; Janssens, K.; Proost, K.; Garcia-Leon, M.;Garcia-Tenorio, R. Characterization of U/Pu particles originatingfrom the nuclear weapon accidents at Palomares, Spain, 1966and Thule, Greenland, 1968.Sci. Total Environ.2007,
376,294–305.Eriksson, M.; Ljunggren, K.; Hindorf, C. Plutonium hot particleseparation techniques using Real-Time Digital Image Systems.Nucl. Instrum. Methods A2002,
488,375–380.Keller, C.The chemistry of the transuranic elements;VerlagChemie GmbH: Weinheim/Bergstr., Germany, 1971; pp 380-392..Holm, E. Review of alpha-particle spectrometric measurementsof actinides.Int. J. Appl. Radiat. Isot.1984,
35,285–290.Hallstadius, L. A. A method for the electrodeposition of actinides.Nucl. Instrum. Methods1984,
223,382–385.Talvitie, N. A. Radiochemical determination of plutonium inenvironmental and biological samples by ion exchange.Anal.Chem.1971,
43,1827–1830.Moring, M.; Ikaheimonen, T. K.; Pollnan, R.; Ilus, E.; Klemola,� �S.; Juhanoja, J.; Eriksson, M. Uranium and plutonium containingparticles in a sea sediment sample from Thule, Greenland.J.Radioanal. Nucl. Chem.2001,
248,623–627.Sill, C. W.; Sill, D. S. Determination of actinides in nuclear wastesand reference materials for ores and mill tailings.Waste Manage.1989,
9,219–229.Croudace, I.; Warwick, P.; Taylor, R.; Dee, S. Rapid procedurefor plutonium and uranium determination in soils using a boratefusion followed by ion-exchange and extraction chromatog-raphy.Anal. Chem. Acta1998,
371,217–225.Komura, K.; Sakanoue, M.; Yamamoto, M. Determination of240Pu/239Pu ratio in environmental samples based on themeasurement of Lx/ X-ray activity ratio.Health Phys.1984,
46,1213–1219.Arnold, D.; Kolb, W. Determination of plutonium content andisotopic ratios in environmental samples by L X-ray andR-particlemeasurements.Appl. Radiat. Isot.1995,
46,1151–1157.Environmental Radioactivity in the North Atlantic RegionIncluding the Faroe Islands and Greenland. 1986; Risø-R-550;RisøNational Laboratory, Denmark,1988.
Environmental Radioactivity in the North Atlantic RegionIncluding the Faroe Islands and Greenland. 1992 and 1993; Risø-R-757(EN); RisøNational Laboratory, Denmark,1997.
Ikaheimonen, T. K.; Ilus, E.; Klemola, S.; Dahlgaard, H.; Ryan,�T.; Eriksson, M. Plutonium and americium in the sediments offthe Thule Air Base.J. Radioanal. Nucl. Chem.2002,
252,339–344.Rollin, S.; Spahiu, K.; Eklund, U.-B. Determination of dissolution�rates of spent fuel in carbonate solutions under different redoxconditions with a flow-through experiment.J. Nucl. Mater.2001,
297,231–243.De Pablo, J.; Casas, I.; Gimenez, J.; Molera, M.; Rovira, M.; Duro,L.; Bruno, J. The oxidative dissolution mechanics of uraniumdioxide, 1. The effect of temperature in hydrogen carbonatemedium.Geochim. Cosmochim. Acta1999,
63,3097–3103.
ES800203F
4722
9
ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 42, NO. 13, 2008